
Fluorescence polarization (FP) is a powerful tool in biochemical research. This method has historically been applied to the study of protein-protein and acid nucleic-protein interactions (Moerke, 2009; Checovich et al., 1926). It is also a suitable method for the screening of drug candidates in the inhibition of enzymes or receptors, such as GPCRs ()Lea & Simeonov, 2011. Rapidly detecting the binding interaction between small molecules and their respective macromolecular targets, FP can easily be applied to the analysis of carbohydrate-lectin associations too (Kakehi et al., 2001).
The technique consists of exciting a fluorescent or fluorophore-labelled molecule with polarised light. Rotational movements of the molecule are responsible for the depolarisation of emitted light, which is monitored by measuring intensity variations in its parallel (I||) and perpendicular (I⊥) components. The angular displacement of light is expressed in terms of anisotropy r or polarisation P. The two quantities are used interchangeably since they are correlated by a simple mathematic equation. Although, the use of anisotropy is strongly encouraged over polarisation since r is normalised by the total of intensity, and it makes calculations easier during the elaboration of the data.
Importantly, the extent of anisotropy is directly proportional to the motion of the fluorescent probe (.Lakowicz, 2006). The rapid Brownian movement of the probe, in turn, is dependent on the viscosity of the solvent and size of the molecule. The applicability of the FP theory in the study of ligand-protein interactions derives from the considerable change of the rate of motion that the fast-moving fluorophore experiences upon binding to its receptor. When the complex is formed, the small ligand molecule is associated with a macromolecule of high mass. Consequently, its tumbling is restricted, and its rotation becomes substantially slower. Therefore, the ligand-receptor pair, after being irradiated, will emit less depolarised light than the free fluorescent ligand (Fig.6).
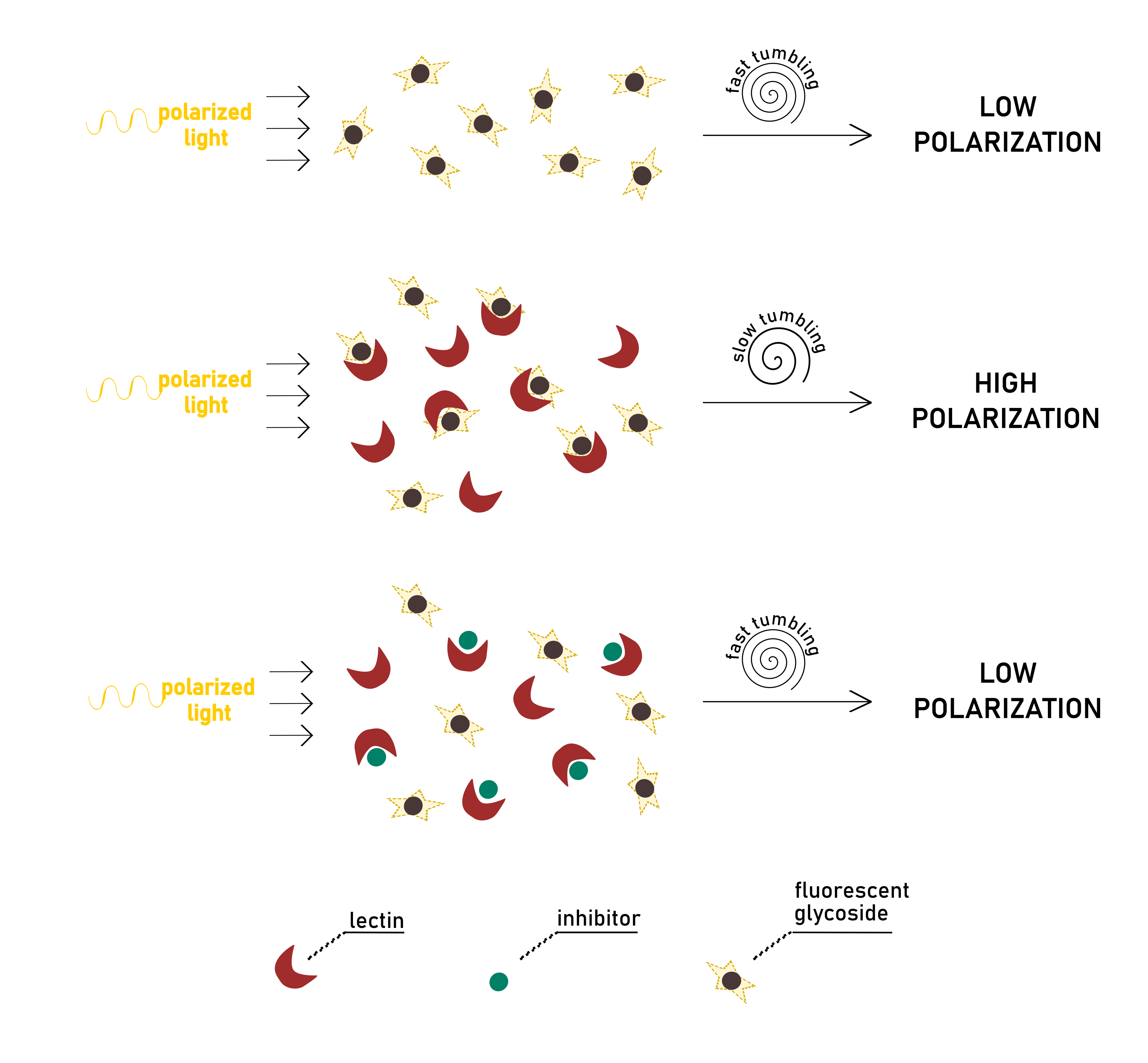
In a typical direct binding experiment, different solutions of protein are added to the fluorescent ligand at a fixed concentration. It is possible to establish a linear proportion between the detected anisotropy and the percentage of bound/free species in solution. After a binding isotherm is generated from the dataset, non-linear regression analysis is applied to obtain quantitative information on affinity constant (Kd).
Alternatively, a competition assay can be established, in which an inhibitor titrates a stock solution of protein and fluorescent ligand. When added to the other components of the mixture, the inhibitor should be able to displace the labelled ligand from the protein binding pocket. The fluorescent probe can, therefore, move freely in solution, lowering polarisation. The signal observed, in this case, is inversely proportional to inhibitor concentration (or, in other words, to the amount of displaced probe). From the resulting binding curve, it is straightforward to extrapolate the value of IC50, as the inhibitor concentration reducing initial protein-probe anisotropy by half. Even for the competitive assay, Kd values for the inhibitor-lectin interaction can be derived. Since total concentrations of biological partners and anisotropy values are known, the inhibitor’s dissociation constant can be calculated directly from each datapoint by solving a set of dependent equations. The details of the calculation can be found in the exhaustive paper of Sӧrme et al (Sorme et al., 2004). Their theories have been successfully exemplified in the study of galectins (Peterson et al. ., 2018; Stegmayr et al., 2016).
Analysis of lectin-carbohydrate interaction by FP is practical and easy, since it does not require immobilisation or washing steps, nor it suffers from severe interferences. Detection is fast, nearly immediate, once the equilibrium of the process under observation is reached. Depending on the kinetics of the interaction, an FP measurement can take just a few minutes. The difficulty in running such an experiment depends mainly on the ease of access to a fluorescent chemical sensor. The first obvious choice in setting an FP assay consists in selecting the proper fluorescent probe. In a competitive binding experiment, the higher the affinity of the fluorescent ligand, the wider the range of inhibitor potencies that can be resolved (Huan, 2003). When measuring the affinity of such inhibitors by direct binding, each compound under evaluation has to be labelled with a fluorophore. Such a strategy has been reported even for the analysis of fluorescent multivalent glycosides, (Li et al., 2014) despite their elaborate synthesis.
Traditionally, fluorescein is the most commonly used agent to label ligands in FP. Fluorescein-labelled monosaccharides have been used in competitive binding assays to evaluate synthetic antagonists of various lectins, such as FilmH, (Han et al., 2010) LecB (Hauck et al., 2013) or BC2L-A (Beshr et al., 2016). Alternatives probes are also available. They can present distinct properties in fluorescence lifetime, quantum yield, extinction coefficient, and stability. One strict criterion for probe selection is its compatibility with the filters for excitation and emission wavelengths installed on fluorescence plate readers. For what concerns the covalent linking of the fluorophore to the ligand of interest, a wide variety of chemistries has been proposed, and procedures are usually standardised. The site of ligation is the primary concern during labelling, in the sense that it must not create an obstacle for binding. Once labelled, ligands should be handled and stored in the dark, since fluorophores are generally sensitive to light and must be protected from it. The FP assay is robust and rarely suffers from interferences. An error that could emerge during analysis is due to non-specific interactions between samples and the surface of the wells. Often this problem can be eliminated by using NBS (non-binding surface) plates that can disrupt such hydrophobic interactions. There is a wide choice for minimising sample consumption and interferences, since FP plates which are commercialised in various materials, well dimensions and sample capacity. The most common are the 96- or 384-well microtiter plates, followed by 1536-well format, which allow a high-throughput screening. The small dimension of the wells is suitable for working with limited volumes. Some plates can reach a lower limit of 3-10 μL per well, as indicated by providers.
Fluorescence polarisation is a sensitive method that allows operating at low concentrations, frequently in the nanomolar range. This is particularly important when proving multivalent glycoside-lectin interactions since low working volumes and concentrations reduce the risk of aggregation and related problems. Additionally, such miniaturised working conditions allow minimising protein and inhibitor consumption (along with costs). The features of the assay permit to screen sets of similar compounds, so to give a preliminary overview over inhibitors’ activities. A few selected compounds can then be chosen for more detailed studies, such as ITC, saving time and materials. At last, the FP technique is not destructive, and the same plate can be read multiple times. This feature is particularly relevant when studying a biological process in its dynamic time-course.