
6.1. Crystallography of Chitins
The availability of chitin in the form of a solid polycrystalline material made it a substrate of choice for X-ray diffraction studies, first on powders and later on oriented fibrillar arrangements. Clark and Smith were the first to make crystal studies of chitin and chitosan using X-ray diffraction.Clark & Smith, 1937 The powder X-ray diffraction patterns recorded from shrimp shells and squid pens exhibit differences that reveal the existence of two polymorphs, namely α-chitin and β-chitin. Rudall & Kenchington, 1973; Blackwell, 1973, Atkins, 1985, Atkins et al., 1979; Saito et al., 1995; Chanzy, 1988 Further structural information was revealed throughout fiber diffraction methods that led the way to the proposal of the occurrence of two structural arrangements that differ in the packing and polarities of adjacent polysaccharide chains.Blackwell, 1973, Gardner & Blackwell, 1975; Minke & Blackwell, 1978 The structure of α-chitin is formed by an arrangement of antiparallel chains, whereas a parallel arrangement of chains forms β-chitin crystals. Further characterizations of the crystalline structure of α-chitin and β-chitin were conducted using electron diffraction experiments on crystalline samp. Persson et al., 1990; Helbert & Sugiyama, 1998 They confirmed the unit cell parameters and space group assignments.
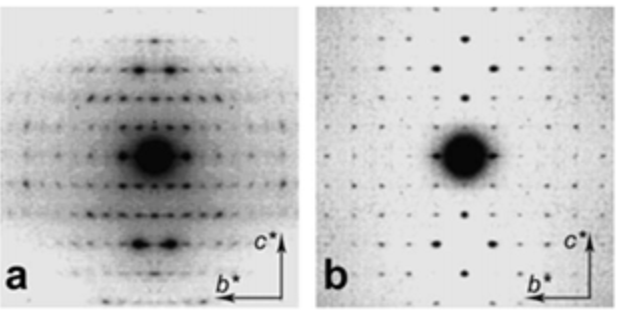
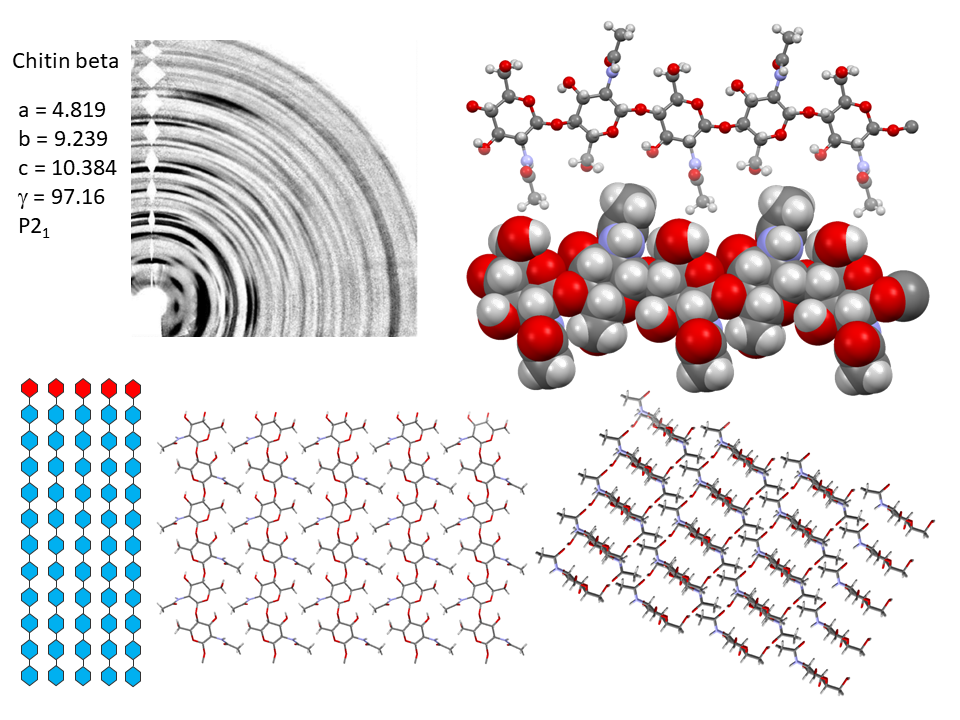
It is only recently that highly accurate determinations of the crystalline structures have been obtained using synchrotron X-ray and neutron diffraction method.Sikorski et al., 2009; Nishiyama et al., 2011; Sawada et al., 2012 While confirming the gross structural features reported previously, these investigations provide the detailed description of the conformations about the glycosidic torsional angles and the resulting twofold helical conformation of the chains. The orientation of the N-acetyl groups, as well as some detailed about the occurrence of disordered orientations of some primary hydroxyl groups, are distinctively characterized. Within the network of intra-chains and inter-chain hydrogen bonds, there is an important involvement of strong C-O…NH hydrogen bonds. In both structures, there is a packing of chitin chains in sheets where intra-sheet hydrogen bonds strongly hold them.
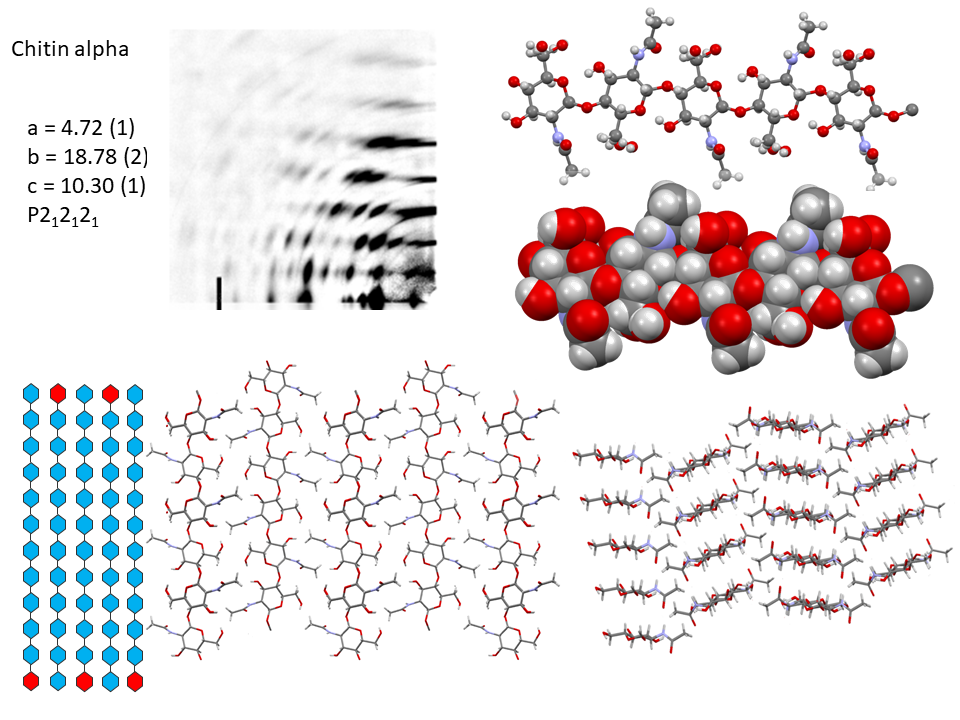
In α-chitin, further hydrogen bonds and van der Waals forces strongly link the sheets together, with the distinct feature displayed by the occurrence of two distinct conformations of the primary hydroxyl groups. β-chitin lacks such a feature, which explains why it is more soluble and more reactive towards solvents and has a greater affinity towards them. The susceptibility to intra-crystalline swelling and the penetration of several polar guest molecules (ranging from water to alcohol and amines) takes place without major distortion of the sheet organization. Once a guest molecule has penetrated the crystalline lattice of β-chitin, another one of a different chemical family can displace it. This feature produces a wide distribution of crystalline β-chitin complexes. However, the reversibility of this swelling and even the crystalline state do not survive in strong acid media typically 6-8 M HCl or concentrated nitric acid.
Nevertheless, removal of the acid restores the crystallinity in the form of alpha-chitin. This process destroys all the beta-chitin crystals and new crystal of α-chitin form during recrystallization. The irreversibility of the α- conversion indicates thataα-chitin is thermodynamically more stable than β-chitin. The fact that α-chitin is always obtained in recrystallization from solution confirms this stability
6.2. Relevance to the Biosynthesis of Chitins
There do exist strong similarities between the polymorphic features observed for chitin and cellulose. In both cases, occurs the irreversibility of the parallel chains to the more stable anti-parallel chains arrangements. However, only in the case of chitin, the two polymorphs occur in native tissues. The crystalline structure of β-chitin (as that of native cellulose) results from a continuous biosynthesis coupled with a chain extension, out of the synthesizing organelle (located in the plasma membrane) and simultaneous crystallization. The case of α-chitin would require a distinct process in the form of a two-step mechanism. The first step would happen in a close organelle (also called chitosome), where it would occur in a somewhat fluid manner. This particular state would allow the chains to reorient and to organize in the most thermodynamically stable antiparallel arrangement. In a second step, the chitosome burst opens, and the microfibrils unravel. At present, such a two-step process works well in vitro. The question remains as to whether such a mechanism operates in vivo and relates to the wide-spread occurrence of alpha-chitin microfibrils in cuticles and not in cocoons where β-chitin occurs.Merzendorfer & Zimoc, 2003 These differences might help to understand the function of chitin in different organisms as significant differences arise between cuticles and peritrophic matrices. As described in the previous section, the architecture of the cuticle results from a hierarchy of structural arrangements starting from α-chitin microfibrils embedded into a protein matrix. By contrast, in peritrophic matrices, the β-chitin microfibrils are generally arranged as a network of randomly organized structures embedded in an amorphous matrix and do not display high ordered arrangements.
6.3. Crystallography of Chitosan and its Polymorphs
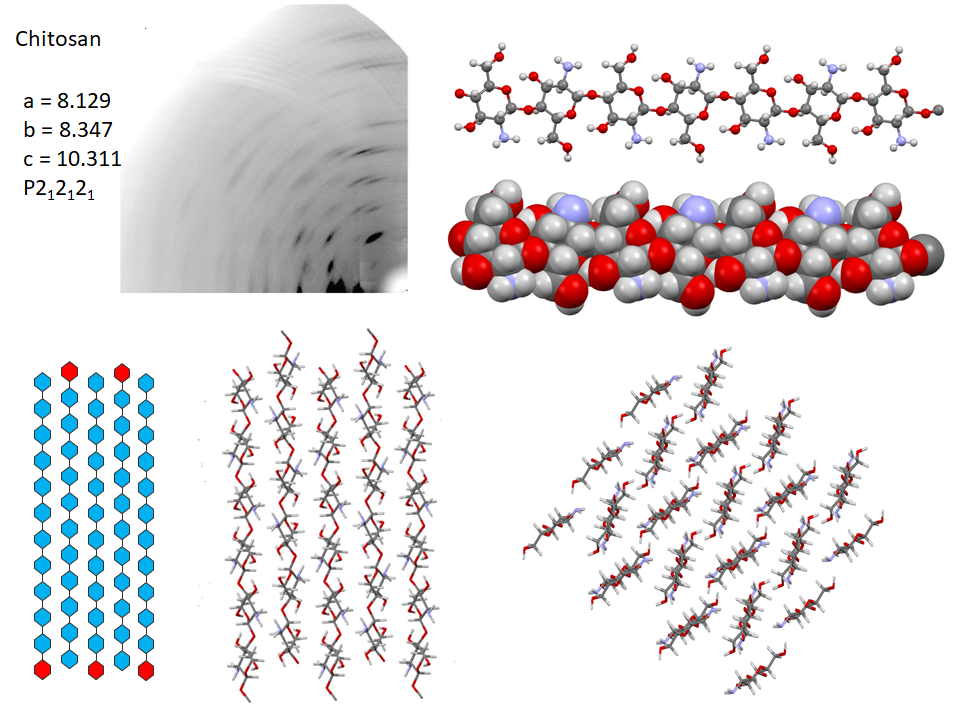
Following the first attempt to characterize the crystalline arrangement of chitosan by X-ray diffraction, the results of many investigations highlighted the influence of experimental conditions : the degree of acetylation, molecular weight, on the crystallinity. The number of reported chitosan crystal structures matches the versatility of chitosan properties themselves. Numerous chitosan complexes resulting from the crystallization of chitosan with acids or transition or post-transition metal ions have been identified, but the detailed characterization of their molecular structures is lacking.Cartier et al., 1992; Ogawa & Inukai, 1987; Ogawa, Oka, & Yui, 1993 In the absence of any ad-duct, chitosan crystallizes at least into two forms : (i) a hydrated orthorhombic P212121 allomorph, proposed with a unit cell containing four molecular chains and eight water molecules.Okuyama et al., 1997 (ii)a highly crystalline anhydrous allomorph, the structure of which has been progressively refined in a series of reports, following successive improvements in preparing and analyzing the crystals.Ogawa et al., 2019
In the most recent X-ray structure determination at atomic resolution, anhydrous chitosan crystallizes as a two-chain unit cell within the P212121 space group.Ogawa et al., 2019 Strictly speaking, this structure corresponds to poly glucosamine (more than “real” chitosan, which by definition, always contains N acetyl Glucosamine residues). The most detailed characterization of the structural feature of anhydrous crystal structure has been obtained using a combination of neutron crystallography and quantum chemical calculation. In the orthorhombic unit cell, the anhydrous chitosan chains adopt a two-fold helical conformation and pack in an antiparallel fashion. The conformational features of the primary hydroxyl groups, their participation in the network of hydrogen bonding network along with the amino groups are unambiguously established. Whereas there is a similarity with the hydrogen-bonding network of cellulose II in helping to stabilize the cohesion between anti-parallel chains. Small differences occur due to the presence of the amino groups that act as poor hydrogen-bonding donors compared to the hydroxyl groups that act as both donors and acceptors
6.4. Solid State Analysis of Chitin and Chitosan
The crystallographic features of α and β chitin and chitosan described above are established on “ideal” samples of well-assessed purity. When it comes to “real” samples, differences in the acetate contents and their distribution, molecular weight, polydispersity and level of crystallinity, generate changes in the overall structure and impact on the properties. Other spectroscopic methods have to be used for analyzing the structure and determining the physicochemical properties of chitin, chitosan, and their derivatives. A series of spectroscopic techniques using X-ray excitation offers an excellent complementarity to Infra-red, NMR, Raman, and UV-Vis analytical techniques.Kumirska et al., 2010
The first level of characterization is performed using X-ray powder diffractometry to identify the nature of the polymorph. The same method provides the determination of the crystallinity index of the samples as well as the degree of N-acetylation of chitin and chitosan. Such X-ray measurements are often used to characterize new derivatives in their processed form or as part as components of materials.
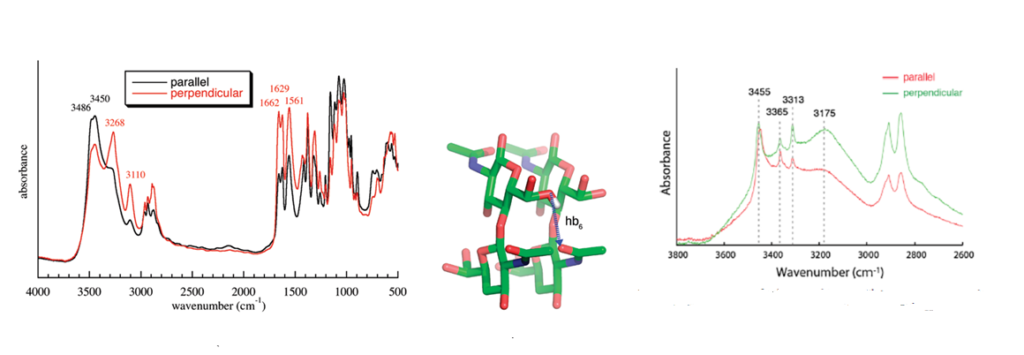
Infrared spectroscopy is and has been widely used to identify the occurrence and nature of chitin and chitosan from different sources.Kumirska et al., 2010 Because of the high crystallinity of the samples, they display a series of very sharp absorption bands, which define specific signatures for α-chitin and β-chitin. Many scientists have published the description and interpretation of the infrared spectra of the two forms of chitin.Darmon & Rudall, 1950, Pearson et al., 1960; Brunner et al., 2009 The C=O stretching region of the amide moiety, between 1600 and 1500 cm-1, is quite interesting as it yields different finger-prints for α-chitin and β-chitin. For α-chitin, the amide I band is split at 1656 and 1621 cm-1, whereas it is unique, at 1626 cm-1 for β-chitin. In contrast, the amide II band is unique in both chitin allomorphs : at 1556 cm-1 for α-chitin and 1560 cm-1 for β-chitin. The availability of the characterized 3D network of hydrogen bonds in α-chitin, explains the main features of its polarized FTIR spectra and sheds new light on the origin of the splitting of the amide I band observed on α-chitin IR spectra. (Sikorski et al., 2009)
The characterization of the solid-state features of chitins and chitosans with a high level of acetylation is most suited to be studied by solid-state 13C and 15N NMR spectroscopy that does not require the dissolution of the polysaccharide. The most crystalline samples yield the best-resolved spectra.Heux, et al., 2000 When recorded at 7.05 T, each spectrum consists of six single-line signals and two doublets at C-2 and C=O, but these doublets are in fact singlets that are split by the effect of the 14N quadrupole coupling. In accounting for this phenomenon, there are therefore only eight signals for the eight carbon atoms of α- and β-chitins. Thus, in both allomorphs, the N-acetyl-D-glucosamine moiety is the independent magnetic residue, in full agreement with the crystal structure of α and β-chitin where this residue is also the independent crystallographic unit. 13C and 15N CP-MAS NMR spectroscopies are also particularly suited to calculating the whole range of acetyl content from 0% to 100% with a small distortion below 5% due to the spectrum baseline and signal broadening. 15N CP-MAS NMR spectroscopy is an instrumental method for calculating the fraction of acetylation FA (or DA).