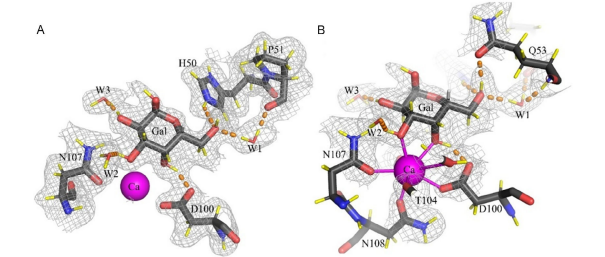
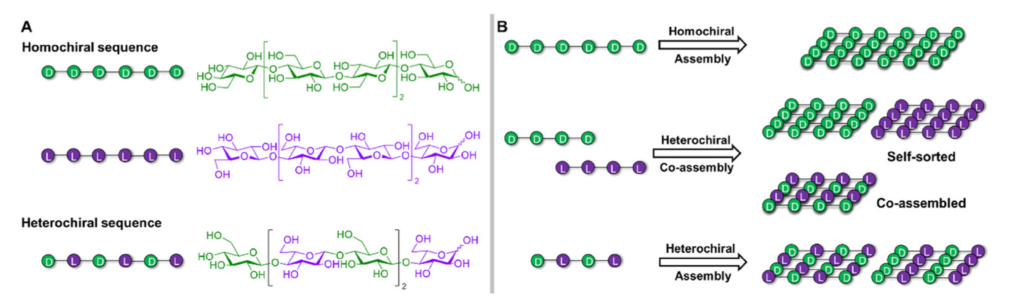
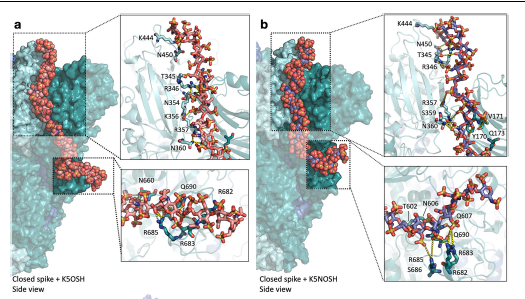
Glycosaminoglycans (GAGs), linear anionic periodic polysaccharides, play pivotal roles in various biologically relevant processes within the extracellular matrix (ECM). These processes encompass cell development, proliferation, signaling, ECM assembly, coagulation, and angiogenesis. GAGs perform their functions through their interactions with specific protein partners, rendering them attractive targets for regenerative medicine and drug design. However, the molecular mechanisms governing protein-GAG interactions remain unclear. Classical structure determination techniques encounter significant challenges when analyzing protein-GAG complexes. This is due to GAGs’ unique properties, including their extensive length, flexibility, periodicity, symmetry, multipose binding, and the high heterogeneity of their sulfation patterns, constituting the “sulfation code.” Consequently, only a limited number of experimental protein-GAG structures have been elucidated. Hence, theoretical approaches are particularly promising in deciphering the code for understanding the structure-function relationship of these complex molecules. In this chapter, the authors focus on the particularities, challenges, and advances of computational methods such as molecular docking, molecular dynamics, and free-energy calculations when applied to GAG-containing systems. These computational approaches provide valuable insights into the enigmatic world of protein-GAG interactions, paving the way for a deeper understanding and potential therapeutic applications.