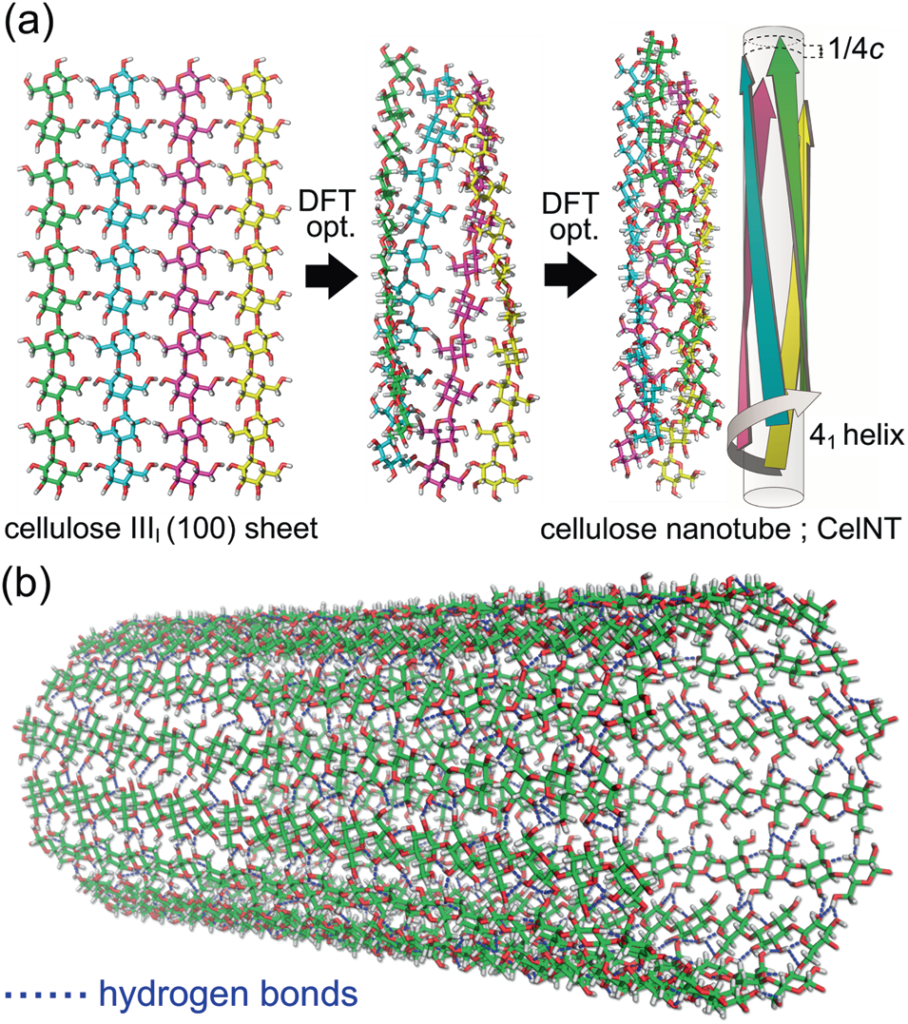
Crystalline polysaccharides are abundant and can be transformed into highly functional materials. However, the molecular basis for forming higher-order structures remains unclear. Computer simulation is an advanced tool for modelling macromolecular structures, and atomistic simulations provide information about crystalline polysaccharides. Fiber deformation, crystalline transition, and novel cellulose nanostructures have been characterized by molecular dynamics simulations and density functional theory calculations of models of molecular chain sheets extracted from the crystal structure of cellulose polymorphs. Extended ensemble molecular dynamics simulations were used to analyze the artificial crystal structure of non-natural amylose analogue polysaccharides, revealing the hexagonal packing of double helices through the self-assembly of molecular chains dispersed in an aqueous solution.
Dissolution simulations of the cellulose and chitin crystalline fibers showed that the anions of the ionic liquids, with their solvating power, played a key role in breaking the intermolecular hydrogen bonds in the crystal structure. The cations, on the other hand, contributed to the irreversible molecular chain dispersion. The good correlation between the solubility of polysaccharides and the predicted number of intermolecular hydrogen bonds prompted the development of a platform combining simulation and machine learning for high-throughput solvent screening of cellulose and chitin.