
Human herpesviruses : Structure and basic properties
Human herpesvirus is a family (Herpesviridae) of large enveloped double stranded DNA (dsDNA) viruses of which eight members have humans as the natural host. The herpesvirus particle has a diameter of about 200 nm and consists of an icosahedral nucleocapsid of 162 capsomers that is surrounded by a double phospholipid bilayer envelope (Mettenleiter, 2002.)
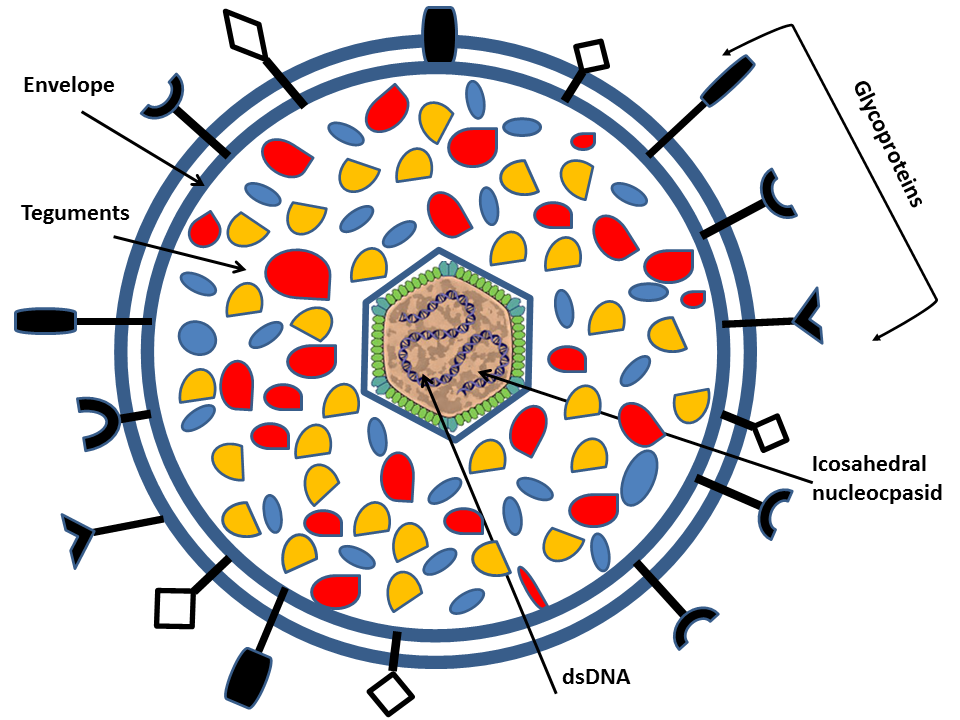
All members have a relatively large genome size ranging from 120kb to 250kb and encode between 70 and 200 genes. Herpesvirus gene expression is strictly regulated starting with expression of immediate early alpha ( α) genes that encodes activators of subsequent viral gene expression as well as proteins that interfere with the cellular antiviral response. This is followed by expression of early ( β) genes encoding proteins important for viral replication (i.e. DNA synthesis) and thereafter, transcription of leaky late (γ1) genes and finally true late (γ2) genes that encode structural proteins including glycoproteins (Fields et al., 2007 Cuchet-Lourenco et al. 2012 Everett et al, 2004). Of special relevance for the present study are the ten or more different glycoprotein species that are located in the envelope of the viral particle, and, later in the infectious cycle, also at the cell surface and other membranes of the infected cell (Mettenleiter, 2002Krummenacher et al, 2013). The organization of these glycoproteins resemble that of normal cellular glycoproteins, but the genetic information for the polypeptide sequence is derived from the viral genome (Fields et al., 2007).
Human herpesviruses, similarities, differences and tropism
Herpesviruses are divided into three subfamilies, alpha- (α), beta- (β) and gamma- (γ) herpesvirinae on the basis of biological properties (Fields et al., 2007). A characteristic feature common to all herpesviruses is that after primary infection – often during childhood- a latent phase is developed that persists lifelong in the infected host. The host reservoir for latent viruses differs dependent on the subfamily belonging of a herpesvirus (Tables 1). Occasionally, latent virus is reactivated resulting in recurrent symptomatic or non-symptomatic productive infections. One explanation behind this coexistence between the herpesviruses and their host is a profound ability of these virus to modulate the host immune response and, in fact, the function of the majority of proteins encoded by each of the large herpesvirus genomes is to interfere and interact with different immune effectors, thereby promoting viral persistence in its host (Goodrum et al., 2012 – Chentoufi &. Benmohamed, 2912 – Daubeuf et al., 2005 – Fakioglu et al., 2008 – Gianella, S., et al., 2012 – Knickelbein et al., 2008).
There are several common characteristics shared by human herpesviruses ; utilization of the same strategy for replication with a strictly controlled transcription program during a productive infection, they encode different glycoproteins that are abundantly expressed both in the viral particle and at the surface of cells during a productive infection, and they can establish and maintain a latent infection by expressing dedicated latency associated transcripts (LATs). Despite these similarities, important differences can be found between the subfamilies and also within each subfamily.
The alpha (α) herpesvirus subfamily contains herpes simplex virus type 1 and type 2 (HSV1 and HSV-2) and varicella zoster virus (VZV) (Table 1a).
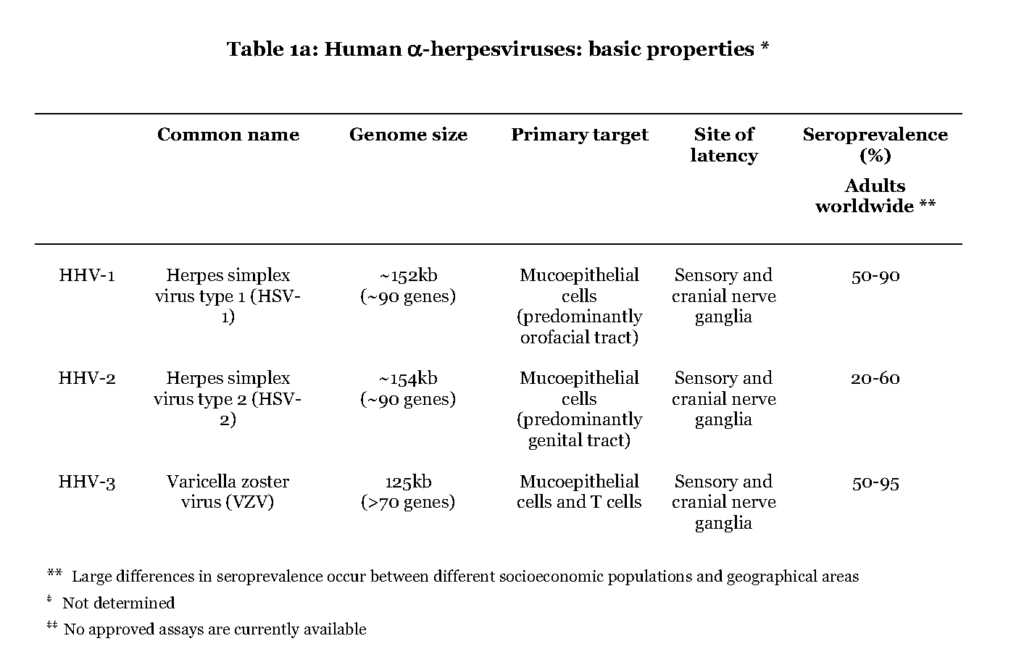
All three cause a primary infection of mucoepithelial cells and later establish a persistent residence in sensory neuronal cells, and they are therefore referred to as neurotropic viruses, a characteristic distinct from the beta (β) and gamma (γ) herpesviruses. Upon reactivation the α-herpesviruses assemble new viral particles that are transported anterograde through the neuronal cell to infect dermal cells (Gilden et al., 2011). The most striking difference between the individual alpha herpes viruses is that HSV1 and HSV-2 cause a local primary infection with a few blisters, cold soars, and with episodes of reactivation occurring essentially at the same location as the first infection, while during VZV primary infection the virus is consecutively spread to local immune cells or cells of the lymphoid system (i.e. white blood cells, leukocytes (Wilson & Mohr, 2012 – Ku et al.,2005). Trafficking of infected T lymphocytes to the skin enables VZV to cause a secondary infection in the form of pox lesions that can cover the entire body (Ku et al., 2002 – Sen et al., 2012). Reactivation of VZV causes a dermatomal-distributed herpes zoster commonly referred to as shingles. Occasionally the α-herpesviruses can also cause severe infections of the central nervous system (CNS). HSV-2-infection can cause primary and recurrent lymphocytic meningitis, while VZV and HSV1 primarily cause encephalitis in case of neurological complications (Steiner et al., 2007 – Landry et al.,2009).
The members of the beta (β) herpesvirus subfamily address a wide range of target cells, including fibroblasts, endothelial cells and leukocytes, in which the virus can replicate and thereby cause a lytic infection (Table 1b).
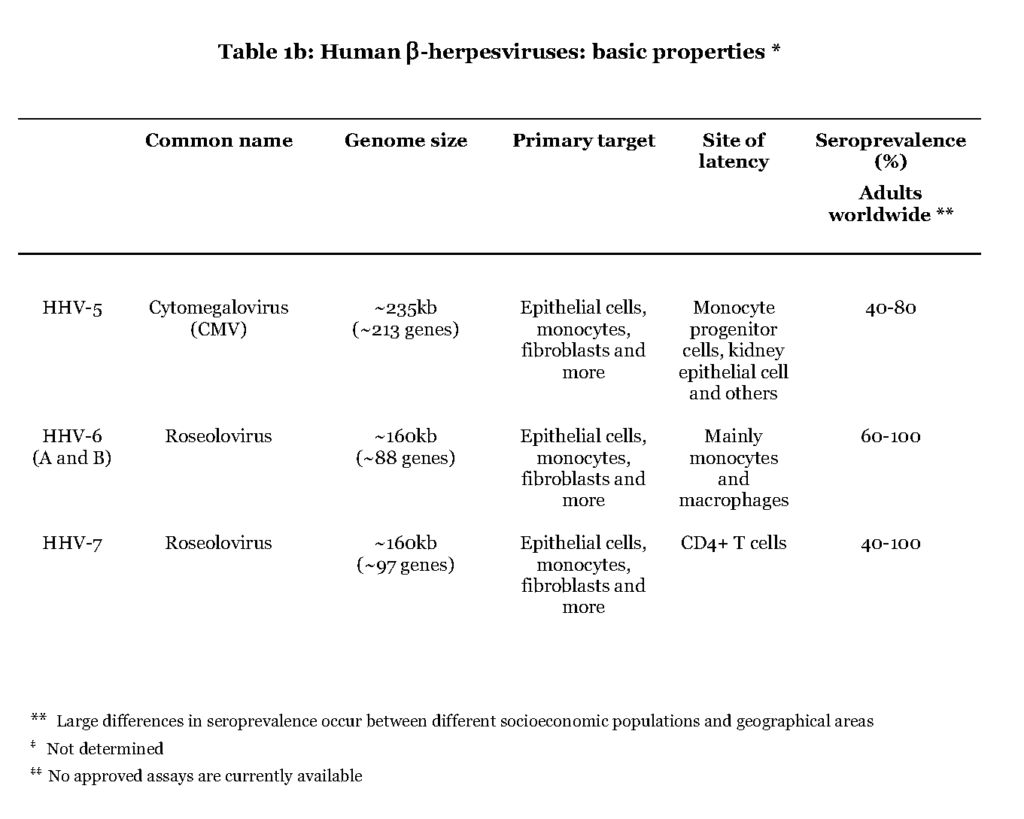
The primary infection by cytomegalovirus (CMV) appears to occur in mucoepithelial cells of the oral cavity although the exact location is not known (Sinzger et al., 2008 – Scrivano et al., 2011). Further viral spread is dependent on white blood cells, leukocytes, and vascular endothelial cells. A subset of leukocytes, myeloid hematopoietic cells (progenitor cells to monocytes), as well as salivary and kidney epithelial cells are believed to be the main reservoirs for harbouring CMV during latency (Seckert et al., 2012 – Cheung et al., 2006). Infections with CMV are asymptomatic or mildly symptomatic, characterized by fever and malaise both during primary and recurrent infectionSteininger, 2007Harvala et al., 2013. The remaining herpesviruses of the beta subfamily share the target cell promiscuity with CMV but the entry pathway for human herpesvirus 6 and 7 (HHV6 and HHV7) is not known. The preferred cell types for establishing a latent infection by these viruses are CD4+ T-lymphocytes, salivary epithelial cells or myeloid lineage hematopoietic cells (White et al., 2012).
The members of the gamma (γ) herpesvirus subfamily include Epstein-Barr virus (EBV) and Kaposi´s sarcoma-associated herpesvirus (KSHV) (Table 1c).
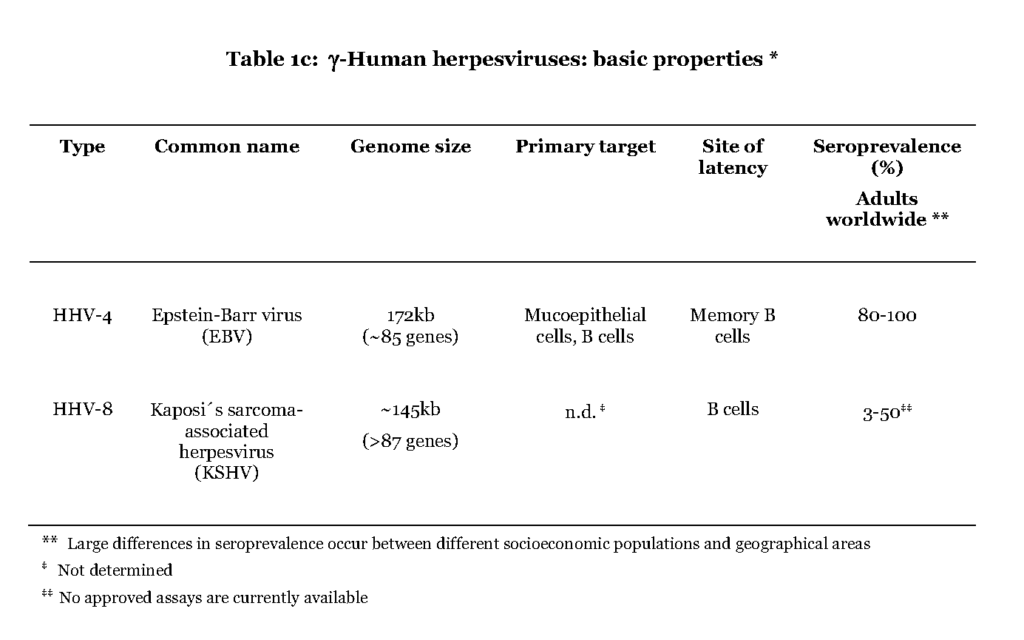
The former is believed to initially infect mucoepithelial cells in the oral cavity while it is not known for the latter. In contrast to the α- and β-herpesviruses EBV and KSHV favour B-lymphocytes for further spread in the host. A distinguishing feature of the primary EBV-infection is that the virus enters the lymphoid tissue of the pharynx and infects resting B cells, which become dividing lymphoblasts. This situation is referred to as infectious mononucleosis (IM) and is characterized by high a number of infected B cells and manifested as fever and swollen lymph nodes (Hislop et al., 2007). The lymphoblasts then differentiate into resting memory B cells, viral transcription is suppressed and low numbers of latently infected cells continue to circulate in the blood (Hislop et al., 2007 – Babcock, et al., 1998). Both EBV and KSHV are associated with lymphoma originating in B cells, after prolonged period of immune suppression, which also separates them from the α- and β-herpesviruses (Cesarman, 2011).
Interplay between the herpesvirus and a functional immune system
The infectious course of herpesviruses can be divided into three different stages starting with acute primary infection, which is followed by establishment of latency and later episodes of reactivation. The acute infection is usually resolved by the concerted action of the innate and adaptive immune response. However, virus is not cleared from the body because of the viral ability to persist in the host despite a functional immune system, which results in a life long symbiotic relationship (White et al., 2012 – Torti & Oxenius, 2012). For example, CMV and EBV as well as HSV1/2 all have evolved means to interfere with the processing steps of major histocompability complex (MHC) class I and II antigen presentation, thereby protecting the infected cell from CD8+ cytotoxic T cells (CTL) and CD4+ T helper cells (Th) respectively (Tortorella et al., 2000). With a functional immune system this complex interplay between the virus and the cells of the immune system ensure that the virus pool is maintained but also confined (Chentoufi et al., 2012 – Hislop et al., 2007 – La Rosa & Diamond, 2012). Consequently, herpesviruses alter their gene expression when establishing a latent infection, suppressing genes important for lytic replication and inducing latency associated transcripts (LATs) also called transcripts expressed in latency (TELs) (Seckert, et al., 2012 – Cheung et al., 2006 – Goodrum et al., 2004).
For a long time a latent herpesvirus infection was regarded as totally dormant, with a quiet viral genome and a resting immune system (Roizman & Sears, 1987). This view is being abandoned as recent reports indicate actively on-going immune responses to various herpesviruses also in asymptomatic individuals (White,et al., 2012 – Sacher et al., 2011).
For example, a large proportion of the T cell pool is directed towards herpesviruses regardless of clinical symptoms and as much as 30% of the total amount of CD8+ T cells have a phenotype towards CMV, EBV and HSV1 even in the absence of an active infection (White et al., 2012 – Sheridan et al., 2007 – Moss & Khan, 2004 – Callan, 2004).
Also, virus-specific CD4+ T cells are important not only for resolving the acute infection but also for controlling latent infection, this pool of CD4+ and CD8+ T cells are constantly surveying the infected cells for markers of infection, specifically epitopes derived from TELs (Cesarman, 2011 – Decman et al., 2005 – Hislop et al., 2007 – Lang et al., 2011 – Nakanishi et al., 2009 – van Leeuwen et al., 2004 – Simon et al., 2006 – Grzimek, 2001). It is now clear that in a subset of the “latently” infected cells the virus frequently reactivates and re-enters a replicative state. Contrary to the picture of quiet viruses that only rarely give episodes of reactivation it now becomes more and more evident that they actually are constantly probing the immune system, causing subclinical infections that are eventually cleared by a functional immune system.
Herpesvirus infections in immune compromised individuals
In individuals with a suppressed immune system the herpesvirus ability to interact with and partially circumvent the immune response becomes a deadly threat. In this respect CMV and EBV are especially problematic, and associated with severe complications primarily as a consequence of primary infection but also during viral recurrence (Kullberg-Lindh et al., 2008 – Fishman, 2013 – Gulley & Tang, 2010 – Kullberg-Lindh et al., 2006). The seroprevalence for herpesviruses is high already early in childhood, increasing with age and reaching almost 100% for EBV and 70% for CMV in adults in developing countries, although large differences occur between socioeconomic populations and geographical areas (Cannon et al., 2010 – Svahn et al., 2006 – Dowd et al., 2013 – Andersson-Ellstrom et al., 1995). As the herpesviruses are so widely spread within the human population the risk of either reactivation or primary infection is extremely high in patients with a defective immune system.
CMV-infections in immunocompromised patients
In patients undergoing transplantation procedures or cancer treatment and in patients with other immune deficiencies (e.g. infection with human immunodeficiency virus – HIV) CMV is a constant menace resulting in frequent episodes of viremia, characterized by a high number of circulating infected leukocytes (Kullberg-Lindh et al., 2008). In solid organ transplant (SOT) patients invasive CMV disease usually occurs within the first year and is most often characterized by fever, weakness, myalgia and myelosuppression (Steininger, 2007). In some patients the infected leukocytes leave the circulation, causing secondary infections that lead to the development of life-threatening end-organ disease, which can affect several organs leading to pneumonitis, hepatitis, carditis, colitis, encephalitis, retinitis and nephritisFishman & Rubin, 1998. Infection with CMV also has indirect effects associated with allograft injury and rejection, increased risk for additional infections and increased risk of EBV-associated post transplant lymphoproliferative disorder (PTLD) (Rubin, 1989).
Due to high incidence of CMV infection (up to 75%) during SOT, the patients are regularly monitored for CMV load in the blood. This is usually done by qPCR with standardized thresholds and once the levels of viral genomes rise above the limit, treatment with antivirals (valgangciclovir and ganciclovir) is started (Asberg et al., 2010). Antiviral treatment, both with ganciclovir and valganciclovir, is effective and the intervention strategy is aimed at only capturing the patients who are at risk of developing CMV disease instead of administering universal antiviral treatment, thereby lowering the risk of emergence of drug resistant CMV strains and reducing the risk of adverse effects of the medication (Owers et al.,2013 – Reusser et al., 1999 – Asberg et al., 2010).
EBV-infections in immunocompromised patients
In most cases the infections with EBV in patients with a suppressed immune system will progress with mild symptoms including fever and malaise but also infectious mononucleosis (IM). However, during the first year after transplantation some develop PTLD, a potential life-threatening neoplasm (Gulley & Tang, 2010). PTLD is a collective name for a diverse group of lymphoproliferative disorders that occurs in 0.3-12.5% of allogenic bone marrow transplantations (BMT) patients and SOT patients, based on the type of organ transplanted and amount and type of immune suppression employed (Allen & Preiksaitis, 2009). PTLD histopathologies include polyclonal lymphoid infiltration where EBV infected leukocytes can be detected in extravasal tissue (Tang et al., 2013). Development of PTLD is highly associated with EBV, and viral genomes are found in over 90% of B cell derived PTLD during the first year after solid organ transplantation (Allen & Preiksaitis, 2009). Also, the incidence of PTLD is higher in adolescent and young transplant recipients than in adults largely explained by their 60-80% sero-negative status (Cesarman, 2011).
In patients with a suppressed immune system EBV also greatly enhances the risk of developing various types of lymphoma that are dependent on expression of transcripts expressed in latency (TELs) and small nuclear RNAs (e.g. EBER1 and 2)(Jochum et al., 2012 – Toczyski et al., 1994). EBV associated lymphomas have a particularly high incidence in HIV infected individuals, in HIV patients with Hodgkin´s lymphoma (HL) almost all cases are associated with EBV and in patients with AIDS and Burkitt lymphomas the association is 40% (Cesarman, 2011). The clinical outcome of PTLD varies ; some lesions diminish after release of immune suppressants, whereas more aggressive treatment might be required, especially after bone marrow transplantation the disease most often follows an aggressive course that in many cases is fatal (Shapiro et al., 1988).
In contrast to the situation for CMV infections, no general antiviral treatment is available for EBV although circumstantial reports suggest that ganciclovir and even ribavirin could be effective (Cohen et al., 2008 – Cohen, 2009). Replication of latent EBV in proliferating B cells does not rely on viral DNA polymerase rendering the ganciclovir type of drugs ineffective. In the absence of effective antiviral treatment several immune modulatory and immune cell therapies have been tried but and the most successful seems to be infusion with EBV-specific cytotoxic T lymphocytes (CTLs) (Louis, et al., 2010 – Heslop, et al., 2010 – Thorley-Lawson & Gross, 2004).
Herpesvirus colonization of extravasal organs – unanswered questions
The most severe complications in both CMV and EBV infections are associated with virus-infected leukocytes leaving the circulation for infiltration of extravasal tissue (Fishman, 2007 – Thorley-Lawson & Gross, 2004). Despite this, the mechanisms behind organ colonization of virus-infected leukocytes remain largely unknown. However, the normal mechanism for leukocyte recruitment to extravasal tissue during inflammation is well characterized and it is possible that herpesviruses utilize the same pathway for colonizing organs. Our group recently published evidence that HSV1, CMV as well as VZV all have the capacity to induce genes relevant for leukocyte transmigration, at least in fibroblasts, supporting this notion (Nystrom et al., 2004 – Nystrom et al., 2007).