Insights from Ab Initio Calculations and Classical Molecular Dynamics Simulations
The study focuses on the conformational properties of biologically relevant monosaccharides belonging to the group of uronates: α-L-iduronate, O2-sulfated-α-L-iduronate, and O2-sulfated-α-L-guluronate, either unfunctionalized or O1-methylated. The authors applied a previously proposed two-step methodology, combining classical MD simulations and subsequent ab initio (QM) calculations performed on a rationally subsampled set of molecular configurations. They found that, regardless of the number of molecular configurations considered, the level of theory,and the weighting scheme applied, none of the QM approaches can predict the correct equilibrium of sulfated iduronates as long as the tight counterion binding is not considered.
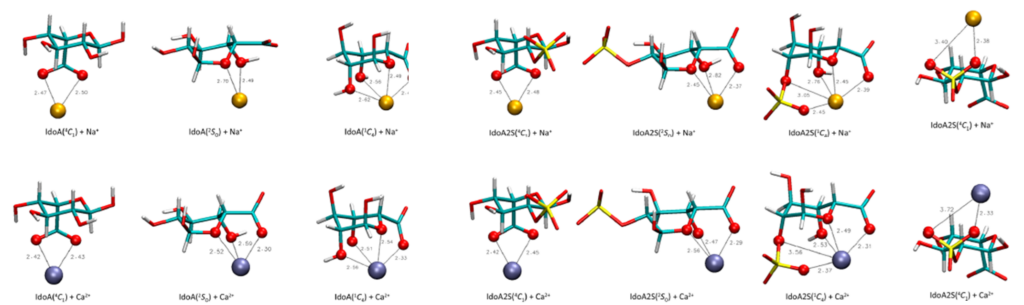
Multicenter, ring-shape-specific binding of either Na+ or Ca2+ ions drastically shifts the conformational equilibrium of the pyranose ring in sulfated iduronates toward 1C4 but does not significantly affect the conformation of non-sulfated compounds. A similar shift is observed upon the protonation of carboxyl groups in all iduronates. In addition, we report a set of average J-coupling constant values related to vicinal protons bound to the pyranose ring of iduronates and corresponding to each of the three main groups of ring conformers, i.e., 4C1, B/S (boat/skew boat), and 1C4. Combined with the conformational energies or experimental data, these values allowed the relative proportions of the ring conformers to be estimated and the Karplus-type equations linking the 3JHH coupling constants to the torsion angles within the pyranose ring to be refined.