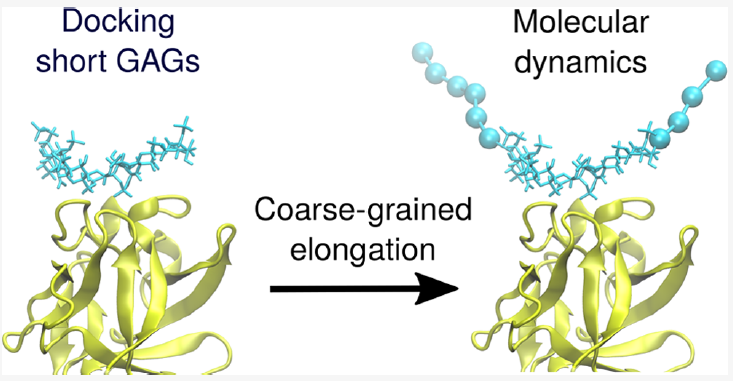
Docking glycosaminoglycans (GAGs) has been challenging because of the complex nature of these long periodic linear and negatively charged polysaccharides. Standard docking tools like Autodock3 are successful when docking GAGs up to the hexameric length. However, they experience challenges to dock longer GAGs properly. Similar limitations concern other docking approaches typically developed for docking ligands of limited size to proteins. At the same time, most of the more advanced docking approaches are challenging for inexperienced users with complex in silico methodologies. In this work, we evaluate the binding energies of complexes with different lengths of GAGs using all-atom molecular dynamics simulations. Based on this analysis, we propose a new docking protocol for long GAGs that consists of conventional docking of short GAGs and further elongation using a coarse-grained representation of the GAG parts not being in direct contact with its protein receptor. This method automated by a simple script is straightforward to use within the Autodock3 framework and is also helpful with other standard docking tools. The authors believe that this method could also be used for docking other linear charged polymers with some minor case-specific modifications.